This post explains how to validate a VCF file prior to performing genomic analyses. This guide will be useful if you want to check if a VCF file is ready for analysis, or if a VCF file is erroneous. This feature has been available from QTLmax 6.0.
Figure 1 shows the “VCF validator” tab selected in QTLmax 6.0.
(Figure 1)
(Figure 2) shows that a VCF file to be validated has been chosen with a project name filled, followed by its VCF validation summary. It is important to not that available types of input files are .vcf or .vcf.gz. This summary is useful to see the current status of a VCF file in terms of:
- whether multi-allelic sites exist.
- whether non-SNP markers exist.
- whether samples without its IDs exist.
- whether missing SNPs exist.
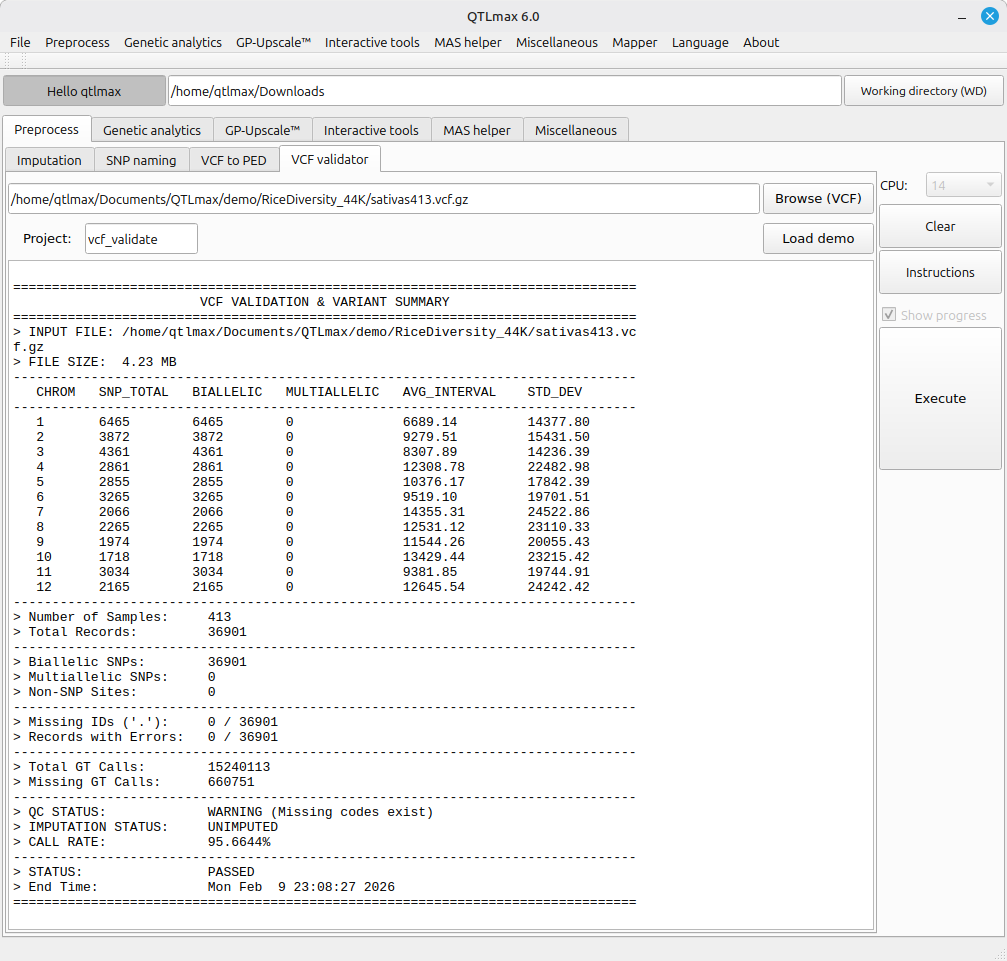
(Figure 2)