Figure 1 shows the tab page for the imputation job. For imputation, you need a file in VCF format or VCF.GZ format (a compressed VCF file). The following settings are required for the imputation job:
(1) Missing ID %: Removes samples with a missing rate higher than the set value (0-1).
(2) Missing SNP %: Removes SNP markers with a missing rate higher than the set value (0-1).
(3) HWE: This refers to the Threshold value for Hardy-Weinberg equilibrium filtering. If there are many zeros after the decimal point, you can use “e” for notation. For example, a value of 0.0000000001 can be expressed as 1e-10.
(4) MAF: Minor allele frequency.
(5) Burn-in: This phase is the initial stage of the Markov Chain Monte Carlo (MCMC) process. The data generated during these rounds is discarded. It is only used to update the model parameters so that the “real” iterations start with a statistically sound foundation.
(6) Iterations: These rounds are sampling iterations that aim at converging on the most likely a SNP code for each missing site.
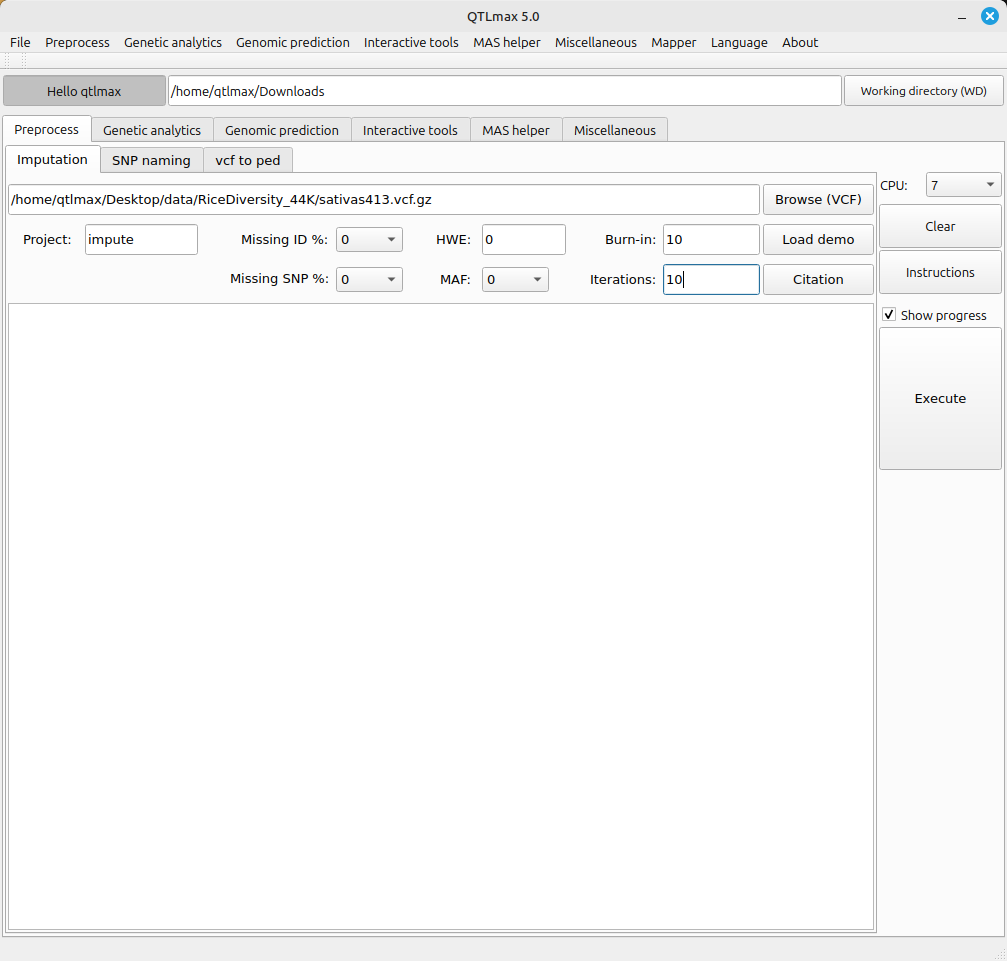
(Figure 1)
After entering all the settings and checking the “Show progress” box, clicking the [Execute] button will bring up a pop-up terminal, displaying the imputation process as it is being performed (Figure 2).
(Figure 2)
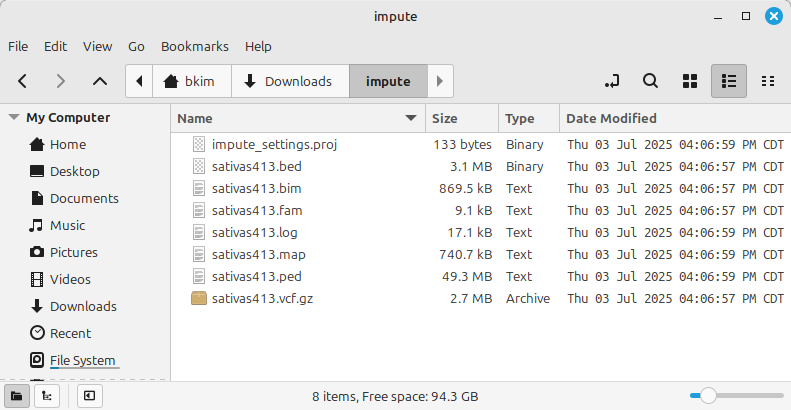
(Figure 3)
Shown in Figure 3 is the imputation result files, which are of three types:
- PLINK format (*.PED, *.MAP)
- PLINK binary format (*.bed, *.bim, *.fam)
- Compressed VCF (*.vcf.gz)
Among the three genomic data formats above, the format required by QTLmax is #1. All three file formats contain the same information and can be interconverted.